Help
1.Introduction
AILDE (Auto In Silico Ligand Directing Evolution) is developed based on the CSO (Computational Substitution Optimization) protocol which was designed to automatically perform computational substitution, energy minimization, and binding affinity evaluation. The method is a new computational approach to substitution-scanning mutagenesis of ligand and could be used as a general strategy of Hit to lead (H2L) optimization in drug and agrochemical design.
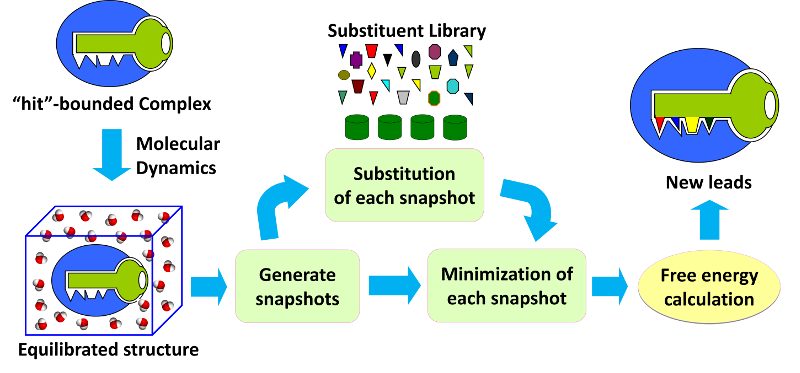
Fig 1. Workflow of CSO
2.Submit Job
- Upload a ligand-receptor complex structure file: A protein-ligand complex file (PDB format, see Notice for more about PDB) is required to upload. The complex structure file can be downloaded from PDB Data Bank. You can also use the docking result as the input complex file, some docking web servers can be used to do the docking job, for example Swissdock, e-LEA3D. Please notice that hydrogens should be deleted before uploading, otherwise the job will give out a mistake.
- Give the RMSF value: The option is used to specify whether to add a MD simulation strategy for the new compound-receptor system. The default is no MD simulation will be added, once value is given, the MD refining step will be performed and the MD simulation will stop until the RMSF of the trajectory is less than the specified value. So the smaller the value, the longer MD simulation time it will cost.
- Name your task: give your job a name, which should be made up letters or numbers. Or you can just ignore it.
- Job password and E-mail address: you can enter a password for your job if you do not want to show your job results to others, you can also ignore it. The E-mail address can be used to send a notice no matter your job is finished or error!
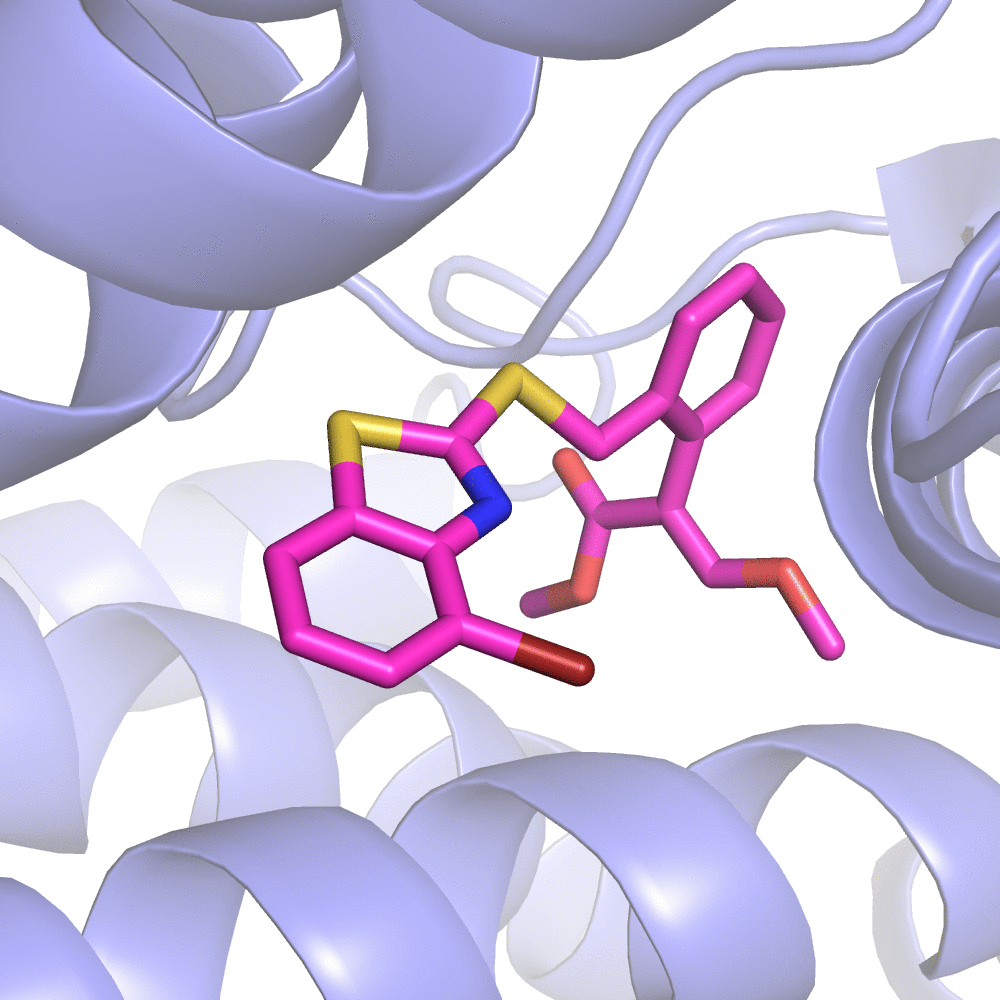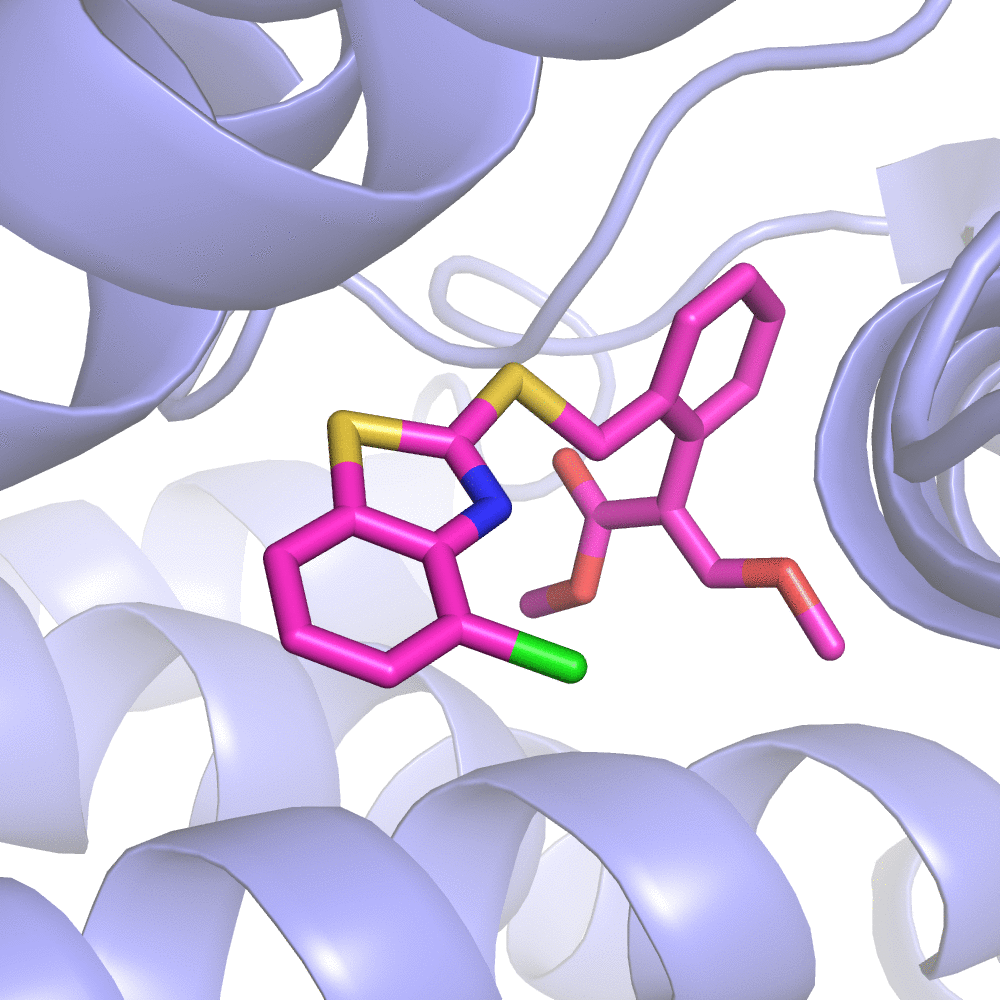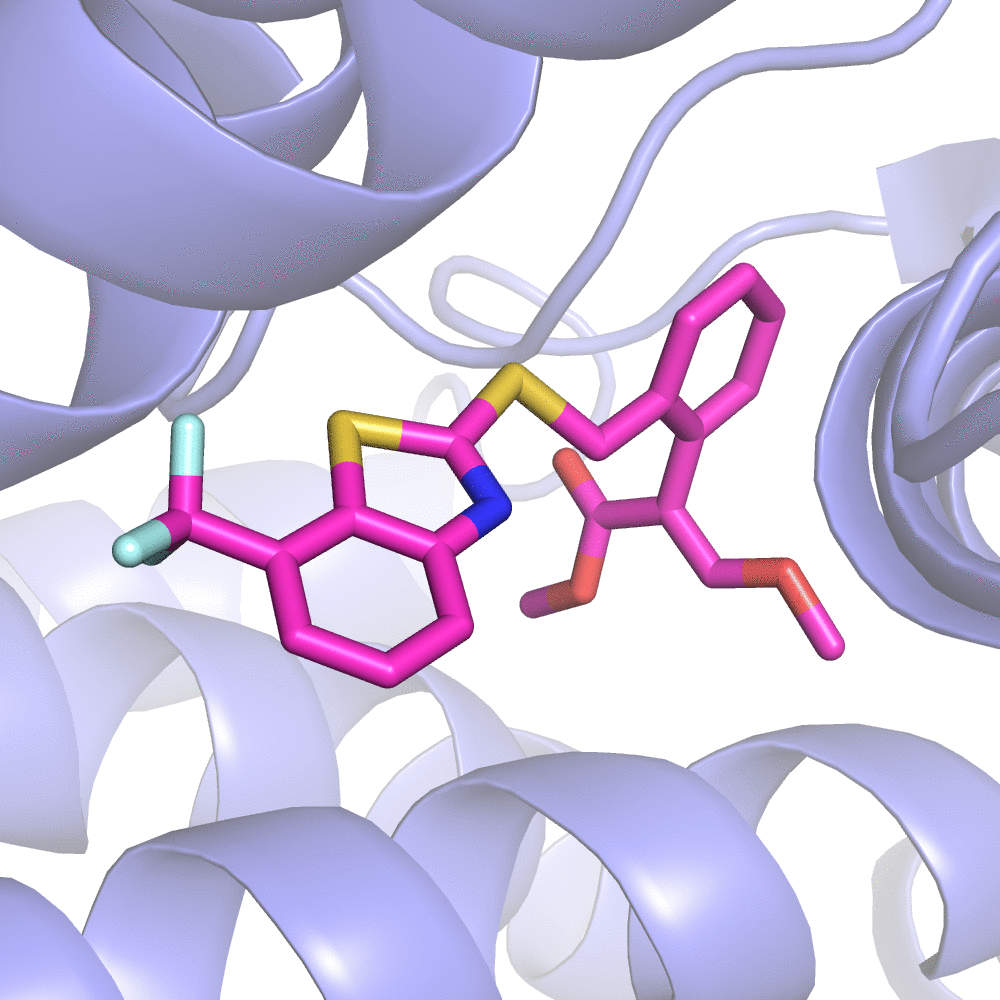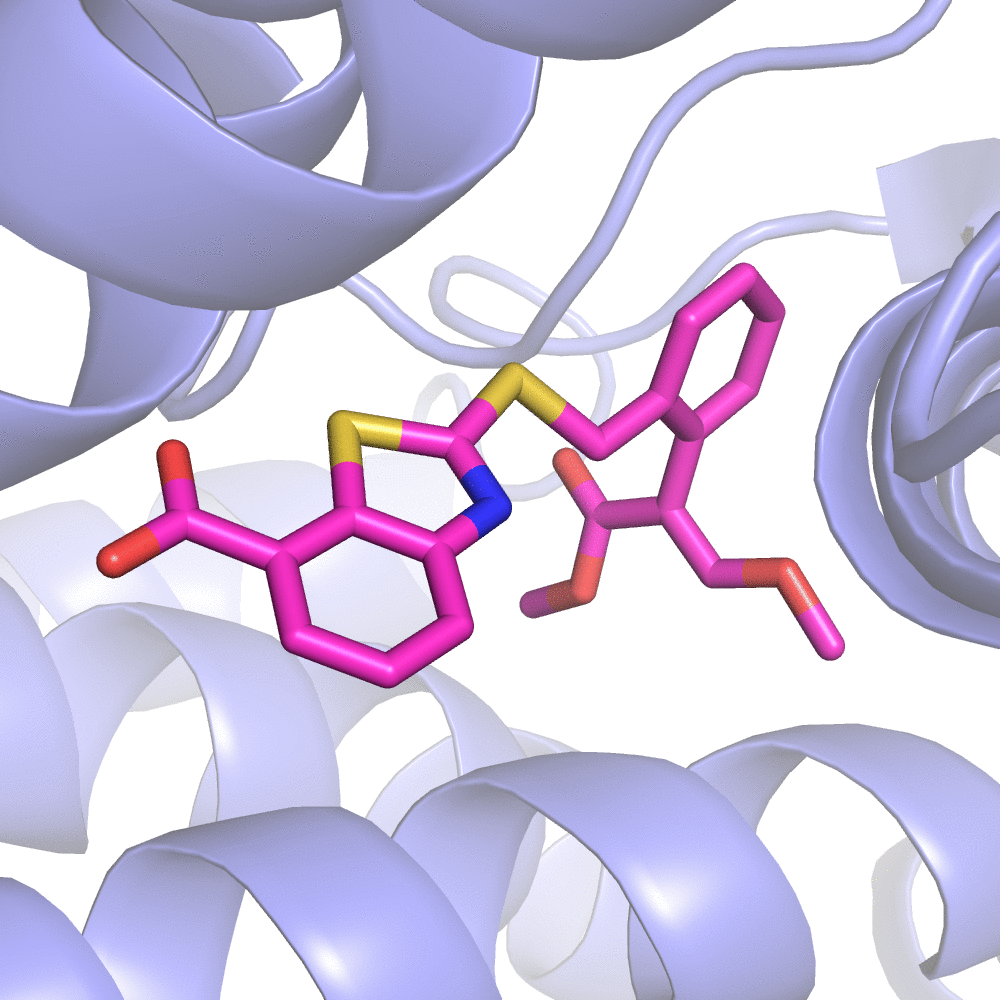
Fig 2. Substitution Scanning Strategy
The server will first RUN the initialization program to obtain all the non-standard residue(s) included in the complex. You can select one as your ligand molecule for Substitution Scanning Optimization.
3.Browse Result
You can click the button to check the status of your job, Running, Queue, Error or Finished. If the job is finished, you can click the ID of your job to access to the login page, and then input your job password to check the result of your job.
4.Notice
Q: What substituents are included in our server?
A: We select 10 small substituent groups for calculation:
NO. Formula Name
01 -Br Bromo
02 -CF3 Trifluoromethylphenol
03 -CH3 Methyl
04 -Cl Chloro
05 -COOH Carboxyl
06 -F Fluorous
07 -NH2 Amino
08 -NO2 Nitryl
09 -OCH3 Methoxyl
10 -OH Hydroxy
Q: What residues can be identified by our server?A: In our server, only 29 residues are supported, the residue names are:
HIS, HIE, HID, HIP:the different protonated states of amino acid Histidine (H).
ALA: amino acid Alanine (A).
GLY: amino acid Glycine (G).
SER: amino acid Serine (S).
THR: amino acid Threonine (T).
LEU: amino acid Leucine (L).
ILE: amino acid Isoleucine (I).
VAL: amino acid Valine (V).
ASN: amino acid Asparagine (N).
GLN: amino acid Glutamine (Q).
ARG: amino acid Arginine (R).
TRP: amino acid Tryptophan (W).
PHE: amino acid Phenylalanine (F).
TYR: amino acid Tyrosine (Y).
GLU, GLH:the different protonated states of amino acid Glutamic acid (E).
ASP, ASH:the different protonated states of amino acid Aspartic acid (D).
LYS, LYN:the different protonated states of amino acid Lysine (K).
PRO:amino acid Proline (P).
CYS, CYM, CYX: the different protonated states of amino acid Cysteine (C).
MET:amino acid Methionine (M).
H2O:water molecule.
Other residues which do not match these names in the complex can be called non-standard residue(s), for example, co-factor(s) that sometimes can be found in a complex strcuture. These non-standard residues will be detected by our server, and generate parameters for them.
Q: What is pdb/mol2 file?
A: pdb: A processible pdb file can be obtained from Protein Data Bank or docking results which should contain ATOM record for protein atoms, HETATM record for non-standard residue (including ligand) and TER record to separate different chains and to mark non-standard residues.For HETATM record and ATOM record, columns 7-11 should be atom serial number, columns 13-16 should be atom name, columns 18-20 should be residue name, columns 23-26 should be residue sequence number, columns 31-38 stand for orthogonal coordinates for X in Angstroms, columns 39-46 stand for orthogonal coordinates for Y in Angstroms, columns 47-54 stand for orthogonal coordinates for Z in Angstroms, columns 77-78 for Element symbol. Generally, pdb files from the protein data bank are always acceptable by our server, but you may need to check your files if you get the pdb file through other ways (e.g. docking, homology modeling).
Here is a pdb file example:
Example:
1 2 3 4 5 6 7 8
12345678901234567890123456789012345678901234567890123456789012345678901234567890
ATOM 3001 N TRP 378 91.533 115.037 126.730 N
ATOM 3002 CA TRP 378 90.600 113.899 126.701 C
ATOM 3003 C TRP 378 89.250 114.198 127.341 C
ATOM 3004 O TRP 378 89.145 114.768 128.432 O
ATOM 3005 CB TRP 378 91.230 112.721 127.449 C
ATOM 3006 CG TRP 378 91.424 111.487 126.659 C
ATOM 3007 CD1 TRP 378 90.925 111.208 125.407 C
ATOM 3008 CD2 TRP 378 92.129 110.319 127.075 C
ATOM 3009 NE1 TRP 378 91.276 109.932 125.027 N
ATOM 3010 CE2 TRP 378 92.025 109.360 126.020 C
ATOM 3011 CE3 TRP 378 92.872 109.981 128.223 C
ATOM 3012 CZ2 TRP 378 92.632 108.084 126.081 C
ATOM 3013 CZ3 TRP 378 93.463 108.696 128.292 C
ATOM 3014 CH2 TRP 378 93.324 107.762 127.223 C
ATOM 3015 OXT TRP 378 88.213 113.721 126.886 O
TER
HETATM 3018 CHA HEM 2 88.420 89.817 143.178 C
HETATM 3019 C1A HEM 2 88.328 88.707 144.014 C
HETATM 3020 NA HEM 2 87.626 88.531 145.174 N
HETATM 3021 C4A HEM 2 87.863 87.326 145.752 C
mol2: A Tripos mol2 file is a complete, portable representation of a SYBYL molecule. It is an ASCII file which contains all the information needed to reconstruct a SYBYL molecule. For more information about mol2 file, please see mol2.pdf for details.
Here is a mol2 file for benzene:
Example:
1 # Name: benzene
2 # Creating user name: tom
3 # Creation time: Wed Dec 28 00:18:30 1988
4
5 # Modifying user name: tom
6 # Modification time: Wed Dec 28 00:18:30 1988
7
8 @MOLECULE
9 benzene
10 12 12 1 0 0
11 SMALL
12 NO_CHARGES
13
14
15 @ATOM
16 1 C1 1.207 2.091 0.000 C.ar 1 BENZENE0.000
17 2 C2 2.414 1.394 0.000 C.ar 1 BENZENE0.000
18 3 C3 2.414 0.000 0.000 C.ar 1 BENZENE0.000
19 4 C4 1.207 -0.697 0.000 C.ar 1 BENZENE0.000
20 5 C5 0.000 0.000 0.000 C.ar 1 BENZENE0.000
21 6 C6 0.000 1.394 0.000 C.ar 1 BENZENE0.000
22 7 H1 1.207 3.175 0.000 H 1 BENZENE0.000
23 8 H2 3.353 1.936 0.000 H 1 BENZENE0.000
24 9 H3 3.353 -0.542 0.000 H 1 BENZENE0.000
25 10 H4 1.207 -1.781 0.000 H 1 BENZENE0.000
26 11 H5 -0.939 -0.542 0.000 H 1 BENZENE0.000
27 12 H6 -0.939 1.936 0.000 H 1 BENZENE0.000
28 @BOND
29 1 1 2 ar
30 2 1 6 ar
31 3 2 3 ar
32 4 3 4 ar
33 5 4 5 ar
34 6 5 6 ar
35 7 1 7 1
36 8 2 8 1
37 9 3 9 1
38 10 4 10 1
39 11 5 11 1
40 12 6 12 1
41 @SUBSTRUCTURE
42 1 BENZENE1 PERM 0 **** **** 0 ROOT
Q: How to consider important water molecule(s)?
A: If you want to take important water molecule(s) into consideration when performing AILDE job, please change the residue name of water molecule(s) into 2HO, because other water molecule(s), always with the name of WAT or HOH will be removed during the calculation.